在微觀的(de)尺度下(xià),人(rén)體就像是一個(gè)沒有硝煙(yān)的(de)戰場(chǎng),除了(le)細菌、病毒這(zhè)些外來(lái)入侵者,還(hái)有來(lái)己方陣營的(de)“叛徒”——癌細胞。衆所周知,癌症是基因突變導緻的(de),但并不意味著(zhe)隻要發生了(le)基因突變就會得(de)癌症,這(zhè)歸功于腫瘤免疫監視的(de)存在。
自20世紀50年代末
免疫監視理(lǐ)論提出以來(lái),免疫系統在限制腫瘤發生中的(de)作用(yòng)已被廣泛接受。這(zhè)一理(lǐ)論的(de)核心是腫瘤細胞由于基因突變而産生了(le)非自身抗原,從而引發免疫反應,因此,免疫系統可(kě)以在臨床表型出現之前不斷識别和(hé)清除這(zhè)些癌前細胞。
然而,這(zhè)一理(lǐ)論無法解釋爲什(shén)麽腫瘤仍然能夠在免疫系統功能健全的(de)人(rén)身上發生發展,也(yě)就是這(zhè)些腫瘤爲何會從免疫監視中逃逸。
癌症免疫編輯理(lǐ)論則認爲,免疫系統和(hé)腫瘤細胞之間的(de)相互作用(yòng)是一個(gè)動态過程,不僅涉及腫瘤預防,而且還(hái)塑造了(le)發展中的(de)腫瘤的(de)免疫原性。具體來(lái)說,識别腫瘤抗原的(de)T細胞介導免疫清除的(de)過程中,免疫系統通(tōng)過影(yǐng)響腫瘤微環境或者引起腫瘤細胞自身改變,誘導形成了(le)免疫原性較低的(de)腫瘤細胞亞群。低免疫原性的(de)腫瘤細胞是導緻免疫監測和(hé)免疫治療最終失敗的(de)根本原因。
盡管這(zhè)一理(lǐ)論爲腫瘤免疫逃逸提供了(le)有說服力的(de)解釋,但關于早期或原始緻瘤細胞發生免疫逃逸的(de)原因仍然不清楚。例如,在免疫系統施加選擇性壓力之前,早期緻瘤細胞在腫瘤抗原表達上是否具有内在異質性?是什(shén)麽機制決定和(hé)調控它們的(de)抗原表達水(shuǐ)平?
2022年2月(yuè)3日,中國醫學科學院基礎醫學研究所黃(huáng)波教授團隊(助理(lǐ)研究員(yuán)呂家迪和(hé)博士後周雅博爲論文共同第一作者)在 Science 子刊 Science Translational Medicine 上發表了(le)題爲:Epigenetic modification of CSDE1 locus dictates immune recognition of nascent tumorigenic cells 的(de)研究論文。
該研究
揭示了(le)早期腫瘤幹細胞躲避免疫監視的(de)謎團,由此從源頭上解釋了(le),爲什(shén)麽有的(de)人(rén)不抽煙(yān)、不喝酒且生活作息均衡但最終還(hái)是患上癌症,而有的(de)人(rén)抽煙(yān)、喝酒、熬夜卻沒有患上癌症——他(tā)們的(de)體内都會不可(kě)避免地出現腫瘤細胞,但隻有一小部分(fēn)腫瘤細胞會能夠逃逸腫瘤免疫監視,最終發展爲癌症。而這(zhè)些細胞的(de)不同逃逸腫瘤免疫監視的(de)能力,
是由CSDE1基因的(de)差異表觀遺傳修飾和(hé)随後的(de)差異表達所決定的(de)。
該研究
揭示了(le)腫瘤免疫逃逸的(de)新機制,對(duì)于腫瘤的(de)早期預防和(hé)免疫治療預後具有重要幫助,也(yě)爲發展下(xià)一代腫瘤免疫治療指明(míng)了(le)新的(de)方向。


黃(huáng)波教授
黃(huáng)波,北(běi)京協和(hé)學者特聘教授,中國醫學科學院基礎醫學研究所副所長(cháng)。國家傑出青年科學基金獲得(de)者,教育部“長(cháng)江學者”特聘教授,國家“百千萬工程”領軍人(rén)才。黃(huáng)波教授緻力于研究腫瘤免疫、腫瘤免疫治療、生物(wù)機械力學、腫瘤休眠、腫瘤代謝等前沿醫學科學問題,在腫瘤免疫逃逸、T細胞耗竭及新型腫瘤免疫療法等領域取得(de)一系列原創性成果。
免疫系統與腫瘤細胞的(de)博弈是高(gāo)度動态的(de),即免疫系統在不斷識别和(hé)清除腫瘤細胞的(de)同時(shí),腫瘤細胞也(yě)會通(tōng)過幹預識别進行免疫逃逸。打個(gè)比方,免疫系統和(hé)癌細胞就像是在玩“捉迷藏”,免疫監視成功與否不僅僅取決于免疫系統的(de)偵察能力,還(hái)取決于腫瘤細胞的(de)反偵察能力。
在人(rén)體内,DNA突變是時(shí)刻發生的(de),但隻有在極少數情況下(xià),DNA突變才會導緻正常細胞轉化(huà)爲腫瘤細胞。然而,即便如此,這(zhè)些腫瘤細胞也(yě)不等同于腫瘤,在人(rén)群中隻有極少數個(gè)體的(de)腫瘤細胞最終發展成爲有臨床症狀的(de)腫瘤,即人(rén)們常說的(de)“得(de)了(le)腫瘤”。那麽,同樣是惡性轉化(huà)的(de)腫瘤細胞,爲什(shén)麽有的(de)會長(cháng)成腫瘤,而有的(de)有不會長(cháng)成腫瘤呢(ne)?
這(zhè)些最終會長(cháng)成腫瘤的(de)極少數的(de)腫瘤細胞,被稱爲
腫瘤幹細胞(Cancer stem cells,CSCs),它可(kě)以逃避免疫監視而作爲腫瘤發生的(de)原始緻瘤細胞,是腫瘤發生的(de)根源。遺憾的(de)是,由于長(cháng)期以來(lái)缺乏研究腫瘤幹細胞與免疫系統相互作用(yòng)的(de)代表性的(de)體内外模型,腫瘤幹細胞發生免疫逃逸的(de)根本性問題仍未闡述清楚。
在這(zhè)項最新研究中,
黃(huáng)波團隊基于生物(wù)機械力學理(lǐ)論建立了(le)
軟三維纖維蛋白凝膠(Soft 3D fibrin gel)培養系統,并利用(yòng)此系統在體外篩選、富集并擴增腫瘤幹細胞,從而獲得(de)由單個(gè)腫瘤幹細胞生成的(de)腫瘤模型。
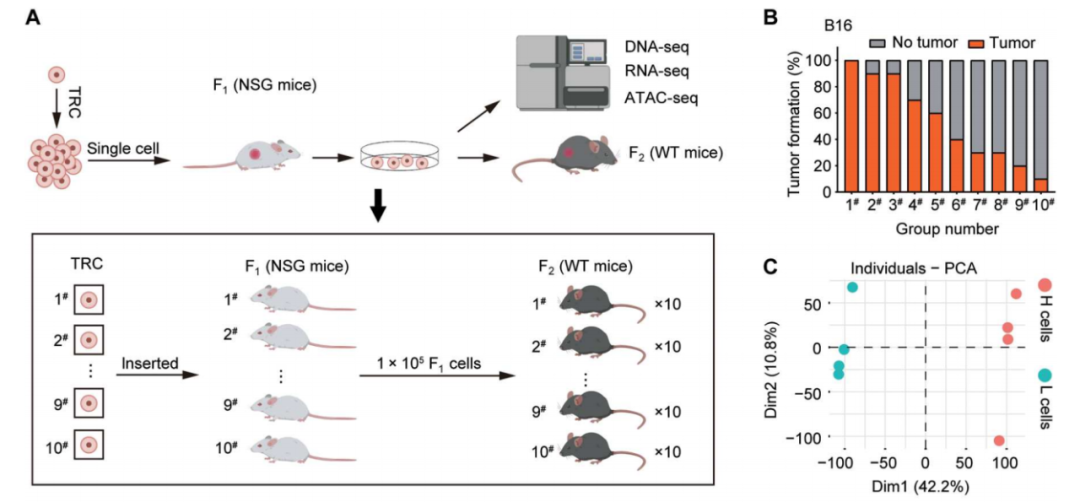
通(tōng)過3D纖維蛋白凝膠培養系統獲得(de)由單個(gè)腫瘤幹性細胞生成的(de)腫瘤模型
研究團隊将這(zhè)類3D培養細胞稱爲
腫瘤再生細胞(Tumor-repopulating cells,TRCs),并通(tōng)過TRCs在免疫缺陷或免疫正常的(de)小鼠中形成腫瘤,以此研究腫瘤微環境對(duì)腫瘤幹細胞的(de)特性的(de)影(yǐng)響。
有趣的(de)是,腫瘤幹細胞的(de)形成不僅受到化(huà)學信号的(de)影(yǐng)響,而且還(hái)受到
生物(wù)機械力信号的(de)調控。既往研究也(yě)表明(míng),腫瘤微環境中生物(wù)機械力在調控腫瘤幹細胞的(de)形成及維持其幹性方面起著(zhe)至關重要的(de)作用(yòng),但尚未清楚其對(duì)免疫逃逸有何影(yǐng)響。
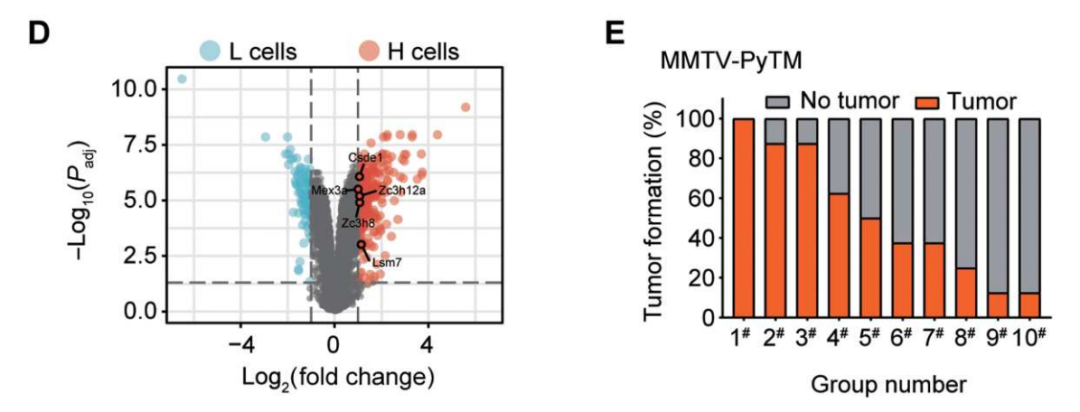
不同的(de)腫瘤幹性細胞具有不同的(de)免疫逃逸能力
黃(huáng)波團隊發現,腫瘤微環境的(de)生物(wù)機械力影(yǐng)響了(le)TRCs中一種名爲
CSDE1的(de)基因的(de)表達,并顯示出幹擾素反應性下(xià)降及抗原呈遞功能減弱的(de)特性。此外,敲除CSDE1的(de)腫瘤幹性細胞隻能在免疫缺陷小鼠體内成瘤,而無法在免疫正常小鼠中成瘤。這(zhè)意味著(zhe)
CSDE1很可(kě)能在腫瘤免疫逃逸中發揮著(zhe)重要作用(yòng)。
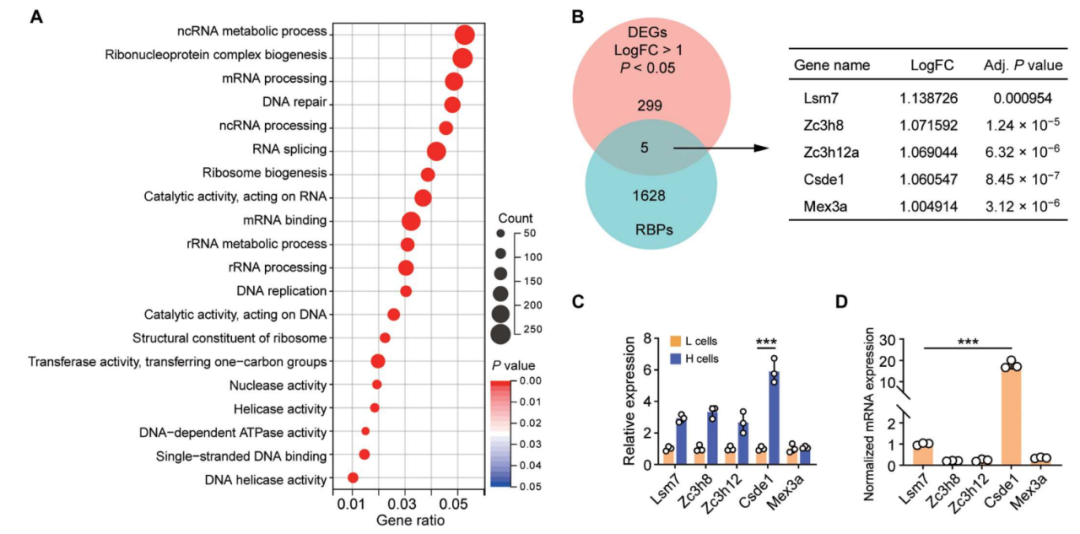
CSDE1調節免疫監視
進一步研究顯示,腫瘤微環境的(de)機械力信号通(tōng)過
SMYD3(一種參與癌細胞增殖的(de)組蛋白甲基轉移酶)表觀遺傳修飾調控
CSDE1,高(gāo)表達
CSDE1的(de)腫瘤幹細胞可(kě)以通(tōng)過穩定T細胞蛋白酪氨酸磷酸酶(TCPTP)的(de)mRNA進而上調其表達量,而TCPTP識别磷酸化(huà)的(de)酪氨酸位點,從而使轉錄激活因子1(STAT1)去磷酸化(huà)而失活。
值得(de)注意的(de)是,活化(huà)的(de)STAT1是啓動抗腫瘤免疫的(de)關鍵信号,促進腫瘤細胞高(gāo)表達腫瘤抗原,增強CD8+T細胞的(de)識别和(hé)殺傷作用(yòng)。因此,一旦STAT1去磷酸化(huà)失活,腫瘤幹細胞獲得(de)了(le)腫瘤免疫逃逸的(de)表型,進而逃避免疫攻擊,最終發展成爲具有臨床症狀的(de)腫瘤。
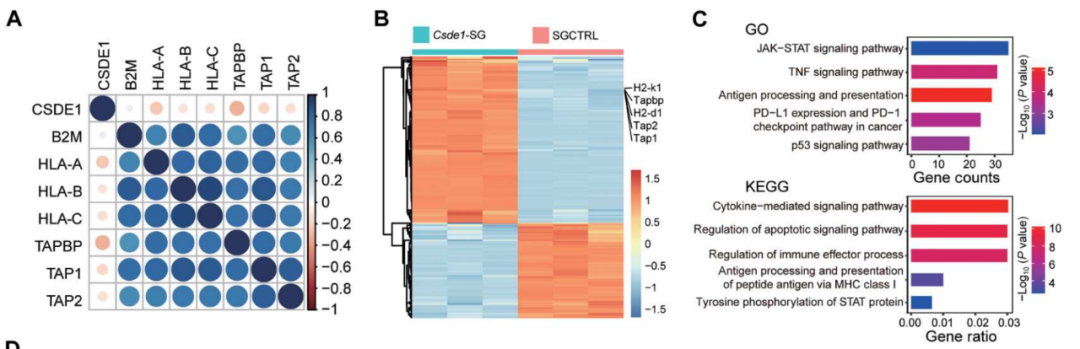
CSDE1通(tōng)過抑制STAT1的(de)磷酸化(huà)來(lái)促進逃避免疫識别
總的(de)來(lái)說,這(zhè)項研究證明(míng)了(le),腫瘤抗原異質性腫瘤細胞的(de)固有特征,在腫瘤發生極早期就已經存在,由表觀遺傳改變引起:機械力信号→SMYD3→CSDE1上調→TCPTP上調→STAT1去磷酸化(huà)失活→腫瘤幹細胞成瘤。
黃(huáng)波團隊從生物(wù)物(wù)理(lǐ)力學角度揭示了(le)腫瘤免疫逃逸的(de)新機制,爲開發新的(de)腫瘤免疫治療及生物(wù)治療策略提供理(lǐ)論基礎,對(duì)于腫瘤的(de)早期預防和(hé)免疫治療預後具有重要作用(yòng)。
中國醫學科學院基礎醫學研究所助理(lǐ)研究員(yuán)
呂家迪和(hé)博士後
周雅博爲論文共同第一作者,中國醫學科學院基礎醫學研究所
黃(huáng)波教授爲論文通(tōng)訊作者。
----------THE END----------
免責聲明(míng):本文系轉載分(fēn)享,文章(zhāng)觀點、内容、圖片及版權歸原作者所有,如涉及侵權請聯系删除!